Benzodiazepines (BZDs), commonly known as “benzo,” are a class of psychoactive drugs widely prescribed for their sedative, hypnotic, anxiolytic, and anticonvulsant properties. These medications exert their effects by targeting GABAA receptors (GABAARs) in the central nervous system, the primary mediators of inhibitory neurotransmission. For over four decades, the GABAA receptor has been recognized as the molecular target of Benzo Gaba action. However, a comprehensive understanding of the precise modulation mechanism at the channel protein level remains an area of active research. Recent advancements in functional studies and, notably, cryo-EM structures of GABAA(α1)2(βX)2(γ2)1 receptors bound with BZDs have provided significant insights, paving the way to bridge this knowledge gap. This article delves into the mechanistic interpretations of functional and structural evidence elucidating how benzo GABA exerts its influence on GABAA(α1)2(βX)2(γ2)1 receptors. Rather than exhaustively detailing every relevant study or dissecting all mutations and perturbations, the focus here is to highlight the overarching mechanistic principles in light of the latest structural revelations regarding benzo GABA interactions.
1. The Prevalence and Paradox of Benzo GABA Use
Benzodiazepines stand as one of the most frequently prescribed classes of psychotropic drugs in contemporary medicine. In the United States alone, approximately 1 in 25 adults received a benzo GABA prescription in 2021 [1]. The therapeutic applications of benzo GABA are extensive, encompassing the treatment of anxiety disorders, panic attacks, insomnia, seizures, muscle spasms, and alcohol withdrawal symptoms [2]. Despite their clinical efficacy, prolonged benzo GABA use is associated with a spectrum of adverse effects, including tolerance, dependence, withdrawal syndromes, and cognitive impairment [3,4]. Furthermore, benzo GABA is frequently implicated in drug-related overdose cases, often in conjunction with alcohol and opioids [5,6]. Notwithstanding these concerns, the pronounced anxiolytic and muscle relaxant properties of benzo GABA underpin their continued widespread use in therapeutics.
At the molecular level, benzo GABA molecules, characterized by a fused benzene and diazepine ring structure, target specific recognition sites on GABAARs. These receptors are pivotal in mediating inhibitory neurotransmission throughout the central nervous system [7]. The binding of the neurotransmitter GABA to GABAARs triggers the opening of an intrinsic ion channel, facilitating chloride ion influx into the neuron. This chloride influx typically hyperpolarizes the neuronal membrane, inhibiting neural signaling and playing a critical role in maintaining the delicate balance between excitatory and inhibitory signals essential for normal brain function. Benzo GABA compounds modulate the GABAAR response to GABA, effectively fine-tuning GABAergic inhibition within the nervous system.
The diverse effects exhibited by different benzo GABA drugs can be broadly categorized based on their interaction with GABAARs. Some act as positive allosteric modulators (PAMs), enhancing GABA’s effects; others are negative allosteric modulators (NAMs), inhibiting GABAergic signaling; and some act as competitive antagonists, blocking GABA’s action without directly altering channel activity [8]. The clinically relevant sedative and anxiolytic effects of benzo GABA are primarily attributed to PAMs, which amplify GABAergic inhibition [2,9,10]. Unless specified otherwise, “benzo GABA” in this article will refer to PAMs. It’s important to note that certain benzo GABA compounds can exhibit PAM or NAM activity depending on the specific GABAAR subtype. This discussion primarily focuses on GABAA(α1)2(βX)2(γ2)1 receptors, for which high-resolution structures in complex with benzo GABA are currently available, offering unprecedented detail into benzo GABA mechanisms.
2. Historical Milestones in Benzo GABA Research
The inception of benzo GABA therapeutics dates back to 1955 with the synthesis of chlordiazepoxide (Librium) by Leo Sternbach at Hoffmann-La Roche [11]. Intriguingly, the discovery of the sedative and anticonvulsant properties of chlordiazepoxide, and shortly after, diazepam (Valium), preceded the identification of their molecular target [12,13]. Both chlordiazepoxide and diazepam received FDA approval in the early 1960s, rapidly replacing barbiturates, which carried a higher risk of severe side effects. Initially termed “tranquilizers,” terms like “anxiolytics” and “anticonvulsants” are now preferred to describe their therapeutic actions.
Despite their widespread clinical adoption, it wasn’t until 1977 that research using radiolabeled [3H]diazepam binding to brain tissue revealed a specific receptor for benzo GABA localized within the central nervous system [14,15,16,17]. Observations that GABA analogs’ binding potency correlated with their ability to alter benzo GABA affinity for its receptor suggested a potential association between the benzo GABA receptor and GABAAR [18]. Using benzo GABA-affinity chromatography, scientists isolated the benzo GABA receptor from brain tissue and confirmed its association with GABAAR by demonstrating high-affinity binding for both benzo GABA and a GABAAR agonist [19,20,21,22]. Analysis of the purified receptor revealed its heteromeric nature, comprising at least two distinct subunits [23]. Peptide sequences derived from the purified receptor were instrumental in screening DNA libraries and identifying the DNA sequences encoding the GABAAR α and β subunits. Co-expression of these subunits in Xenopus oocytes resulted in the formation of functional GABAARs [24,25,26]. Subsequently, a third subunit, γ, was cloned and found to be essential, alongside α and β subunits, for benzo GABA binding and sensitivity [27].
Further research unveiled multiple variants of the α (1–6), β (1–3), and γ (1–3) subunits [28], the combinations of which dictate the differential effects of benzo GABA [29,30,31,32,33,34,35,36,37,38,39,40,41]. The cloning of GABAAR subunits revolutionized the study of benzo GABA mechanisms, enabling mutagenesis studies to probe the molecular underpinnings of benzo GABA modulation. These studies have pinpointed not only the binding site but also regions crucial for the drug’s modulatory actions. Recent breakthroughs in cryo-electron microscopy (cryo-EM) have enabled the high-resolution structural determination of heteromeric GABAA(α1)2(βX)2(γ2)1 receptors in complex with benzo GABA [42,43,44,45,46,47,48]. Collectively, these functional and structural insights are propelling the ongoing quest for a complete molecular understanding of benzo GABA’s mechanism of action on GABAARs.
3. The Canonical Extracellular High-Affinity Benzo GABA Binding Site
The (α1)2(βX)2(γ2)1 subtype represents the most prevalent GABAAR isoform at synapses [49]. Notably, GABAARs containing α4- and α6-subunits are insensitive to benzo GABA modulation [38]. Comparative analysis of α1 with α6 subunits revealed a critical histidine residue present in α1–3 and 5 subunits, essential for high-affinity benzo GABA binding, which is replaced by arginine in α4 and 6 subunits [50]. Swapping this single residue can either confer (histidine) or abolish (arginine) high-affinity benzo GABA PAM binding for both α1 and α6 subunits [50,51]. Knock-in mice carrying this histidine-to-arginine mutation in α1 are insensitive to the sedative effects of diazepam, although they still exhibit other anxiolytic and motor-impairing effects, possibly mediated by receptors containing α2, 3, or 5 subunits [29,30,31,52].
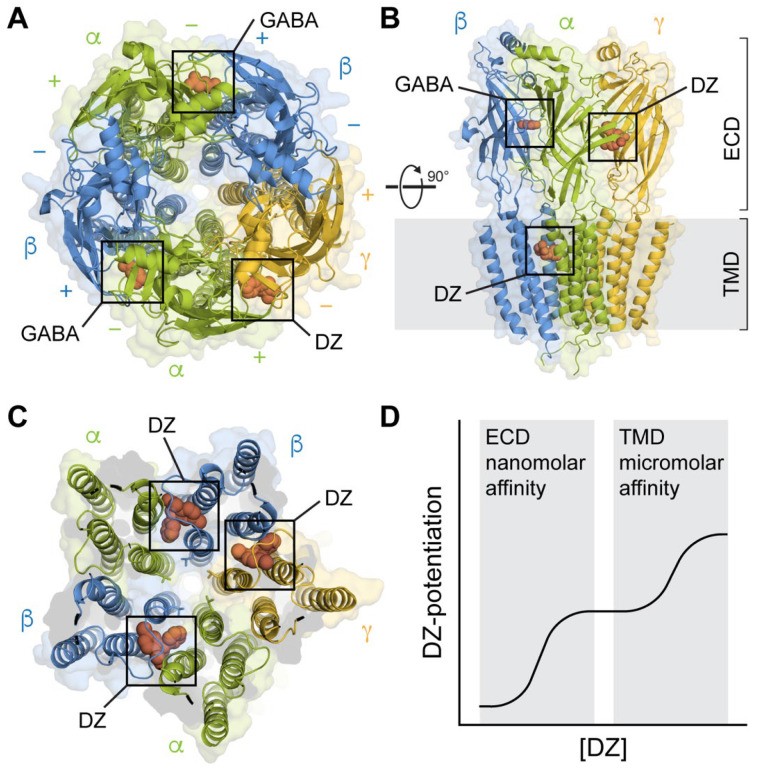
Mutagenesis and photoaffinity labeling studies localized the high-affinity benzo GABA binding site to a region within the extracellular domain (ECD) at the α+/γ− subunit interface. Mutations in this region frequently impaired benzo GABA binding or modulation of GABA-evoked responses [34,53,54,55,56,57,58,59] (Figure 1A,B). Cryo-EM structures have definitively confirmed this region as the primary recognition site for benzo GABA [42,44,45,46,47,48]. This site exhibits homology to the two GABA binding sites located at the β+/α− subunit interfaces. The benzo GABA ligand binds within a pocket situated behind loop C, interacting with residues from both the α and γ subunits (Figure 2). The rings within the core benzo GABA structure engage in π-stacking interactions with surrounding aromatic amino acid residues. The aforementioned critical histidine residue is positioned towards the back of the binding pocket, where it forms a hydrogen bond with the chlorine atom in PAMs like diazepam and alprazolam (Xanax), the fluorine atom in the antagonist flumazenil, or the twin methoxy groups of the NAM DMCM (Figure 2i). Z-drugs, such as zolpidem (Ambien), which share similar sedative and hypnotic effects with benzo GABA, also bind at this canonical site [48,60].
Figure 1.
Structures of synaptic GABAARs in complex with the benzo GABA PAM diazepam (DZ) at both high-affinity ECD and lower-affinity TMD sites. (A) Top-down view with GABA bound at the β+/α− interfaces and DZ bound at the α+/γ− interface in the ECD. Cryo-EM map of GABAA(α1)2(β3)2(γ2)1 from PDB 6HUP. (B) Side view of the structure in A, additionally showing one of several binding sites for DZ in the TMD. Only three subunits are shown for clarity. (C) Same perspective as in (A) for a slice through the TMD. Cryo-EM map of GABAA(α1)2(β2)2(γ2)1 from PDB 6X3X. Three binding sites for DZ are highlighted at the β+/α− and γ+/β− intersubunit pockets between the TM helices and below the M2-M3 linker of one of the subunits. The central pore-gate-forming 9′ leucine residues are shown as sticks near the middle of the pore-lining M2 helices. (D) Biphasic modulation by DZ at the canonical high-affinity site in the ECD and lower-affinity sites in the TMD.
Figure 2.
Canonical ECD binding α+/γ− interface, with diazepam (DZ) shown as white sticks bound behind loop C. Other loops implicated in binding or intersubunit interactions are indicated. Cryo-EM map for GABAA(α1)2(β3)2(γ2)1 from PDB 6HUP. Features discussed in the main text are indicated with spheres for residue Cα atoms and dashed lines for cross-linked residue pairs (numbered labels are referenced in the main text). The critical histidine residue in the β4−β5 loop within the benzo GABA binding pocket (i) and the central valine residue in the α1 M2-M3 linker (viii) are shown as sticks.
4. Transmembrane Binding Sites and Lower Affinity Benzo GABA Interactions
The benzo GABA diazepam (DZ) exhibits a biphasic concentration–response relationship, indicative of both high nanomolar and lower micromolar affinity sites of action (Figure 1D). While mutations in the canonical ECD site discussed above typically affect the high-affinity response, residues within the transmembrane domain (TMD) have been shown to be crucial for the lower-affinity response [61]. Furthermore, introducing mutations in the TMD of ρ1 subunit homomeric GABAARs, which lack a native benzo GABA binding site, can confer benzo GABA sensitivity in the micromolar range. This sensitivity mirrors the low-affinity response observed in heteromeric (α1)2(βX)2(γ2)1 receptors. Cryo-EM structural maps of GABAA(α1)2(βX)2(γ2)1 receptors exposed to high micromolar concentrations of DZ reveal DZ binding at several TMD interfaces between neighboring subunits [45,46] (Figure 1C). Intriguingly, these transmembrane (TM) sites overlap with the binding sites for anesthetics such as propofol [62]. Mutations in key residues within these sites, known to be important for anesthetic binding, also impair the low-affinity action of benzo GABA drugs like DZ and midazolam [63].
It is crucial to recognize that biphasic concentration–response relationships, as observed with DZ, and binding to anesthetic sites in the TMD are not universal characteristics of all benzo GABA compounds. For instance, structures in complex with alprazolam or flumazenil do not exhibit binding in the TMD [45,46]. The effects of flurazepam are insensitive to mutations in these TMD sites [64], and flurazepam potentiation displays a bell-shaped concentration–response curve [65]. Nevertheless, the relevance of anesthetic-like sites of action in the TMD has been established for certain benzo GABA drugs, including DZ and midazolam [63], and warrants further investigation for a broader range of benzo GABA compounds.
5. Exploring Alternative Benzo GABA Binding Sites
Beyond the canonical ECD site at the α+/γ− interface, DZ can also bind to a homologous site at the β+/γ− subunit interface in receptors lacking an α subunit. However, the physiological relevance of such receptor isoforms in neuronal function remains uncertain [66]. The compound CGS 9895, which acts at the canonical α+/γ− site, has also been shown to bind to the homologous α+/β− ECD interface in αβ receptors lacking a γ subunit [65,67]. The ability of these ligands to bind to alternative intersubunit interfaces expands the potential for developing novel subtype-specific modulators of GABAARs [68,69]. However, the significance of these alternative sites as therapeutic targets is yet to be fully elucidated.
6. Benzo GABA Modulation of GABA Binding Affinity
Benzo GABA binding to the classical ECD site does not directly induce channel opening. Instead, it modulates the ability of agonists like GABA to evoke pore opening. The enhanced GABAergic inhibition resulting from benzo GABA PAMs largely originates from their potentiation and prolongation of GABAAR postsynaptic currents [2,9,10]. Notably, benzo GABA potentiation of current responses is observed only with subsaturating GABA concentrations, not saturating concentrations, leading to a leftward shift in the GABA concentration–response curve. This observation suggests that the underlying mechanism involves an enhancement of GABA binding affinity [70,71,72,73,74]. At lower, subsaturating GABA concentrations, where not all GABA binding sites are occupied, enhanced GABA binding effectively leads to larger current responses, comparable to those elicited by higher GABA concentrations. Conversely, at saturating GABA concentrations, where all sites are already occupied, enhanced binding provides no further increase in current. Slower GABA unbinding, due to stabilization of the GABA-receptor complex, can also explain the prolongation of current decay observed with benzo GABA. Consistent with this, studies have shown that DZ primarily affects the frequency of channel opening, which depends on GABA binding rates, with minimal impact on the duration of each opening event, which is governed by pore closure energetics downstream of initial binding events (but see [72] and [70,75]).
For benzo GABA to influence GABA binding, crosstalk between GABA and benzo GABA binding sites is essential. Enhanced binding of radiolabeled DZ in the presence of GABA in brain membranes supports the idea that agonists and benzo GABA PAMs are co-stabilizing [76,77]. Studies examining the rate of modification by methanethiosulfonate (MTS) reagents of introduced cysteines in GABA or benzo GABA ECD sites indicate that ligand binding at either site induces distinct structural changes at the other site [78,79]. The dependence of these structural changes on the nature of the bound ligand (agonist, PAM, antagonist, NAM) suggests their relevance to ligand action and indicates that GABA increases the accessibility of the benzo GABA binding pocket to aqueous reagents.
In all ligand-gated ion channels, agonists that open the channel exhibit higher binding affinity for open (conducting) conformations compared to closed (non-conducting) states. Therefore, any perturbation, such as benzo GABA binding, that increases the probability of channel opening will inherently increase the receptor’s apparent affinity for GABA, even without directly altering affinities for distinct closed and open states. This complexity can significantly complicate the interpretation of equilibrium binding measurements [80,81]. While benzo GABA clearly induces shifts in apparent GABA affinity, the extent to which this reflects direct changes in binding affinities versus shifts in the closed–open equilibrium remains an open question.
7. Benzo GABA Modulation of Pore Gating Mechanisms
Although some single-channel observations align with the idea that benzo GABA primarily modulates GABA binding [72], other studies have reported alterations in macroscopic desensitization or single-channel open durations, which are inconsistent with solely affecting agonist binding [75,82,83]. Furthermore, two lines of evidence strongly suggest that benzo GABA influences channel gating steps occurring downstream of GABA binding. First, peak current responses to saturating concentrations of partial agonists are potentiated by benzo GABA [84,85,86,87]. Partial agonists, while binding at GABA sites, elicit smaller current responses compared to full agonists like GABA. However, at saturating concentrations, all sites are fully occupied. Therefore, the increased maximal current response conferred by benzo GABA must reflect modulation of gating conformational changes in the partial agonist-bound receptor, independent of any effects on the binding step itself.
Second, benzo GABA can directly gate mutant receptors even in the absence of GABA [84,85,86,87,88]. Introducing a mutation in the pore gate can result in channels that spontaneously transition between closed and open conformations [89,90]. The closed–open equilibrium of such spontaneously active mutants is modulated by benzo GABA alone, demonstrating an effect that is independent of GABA binding. While these observations require receptor mutations that significantly alter resting equilibrium, it’s plausible that the mutant channels still gate via the same fundamental mechanism as wild-type channels. Supporting this notion, single-channel analysis of mutants (and their combinations) that confer spontaneous unliganded gating in homologous nicotinic acetylcholine receptors (nAChRs) showed that these mutants alter the closed–open equilibrium independently of agonist-elicited gating [91]. This suggests that the chemical energy derived from benzo GABA binding does influence the pore gate, although the energy may be insufficient to induce appreciable channel opening without additional energy input from agonist binding or other perturbations like mutations.
8. Benzo GABA’s Role in Modulating Closed-Channel Pre-Activation
Single-channel gating dynamics for full versus partial agonists at homologous nAChR and glycine receptors (GlyRs) can be explained by a mechanism where agonist binding promotes a “flipped” or “primed” pre-active conformation of the receptor that precedes pore opening [92,93,94,95]. This model differentiates between partial and full agonists based on their ability to bias the receptor towards this pre-active intermediate state, after which pore opening/closing becomes ligand-independent.
Intriguingly, assuming benzo GABA influences a similar “flipping” or “priming” pre-activation step in GABAARs [96], seemingly conflicting observations supporting benzo GABA modulation of either agonist affinity or pore gating can be readily reconciled [97,98,99]. By modulating the probability of receptor pre-activation, benzo GABA regulates the time spent in a state from which channel opening is possible. Under conditions where pre-activation is not highly probable, enhanced pre-activation by benzo GABA increases the frequency of channel opening. This explains benzo GABA potentiation of currents evoked by partial agonists or subsaturating GABA concentrations where only one of two agonist sites is occupied. Conversely, in saturating GABA conditions with both sites occupied, the channel is already strongly biased towards its pre-active state, and further enhancement of pre-activation by benzo GABA has minimal additional effect on channel opening. This explains the lack of potentiation of currents evoked by saturating GABA. A similar mechanism can account for the modulation by non-benzo GABA drugs like zolpidem, which binds at the canonical benzo GABA site [100].
Given its explanatory power, such a mechanism, where benzo GABA modulates channel pre-activation, is highly plausible for explaining many observed effects of benzo GABA binding in the ECD. However, direct evidence for this mechanism in GABAARs remains lacking. In GlyRs and nAChRs, the pre-active state preceding pore opening is very short-lived. A similarly transient non-conducting intermediate in GABAARs poses challenges for both functional and structural observations. Moreover, observations that single-channel open durations are altered by partial versus full agonists or benzo GABA are not explained by changes in pre-activation alone, which primarily affects opening frequency [75,101]. Some of these complexities may arise from benzo GABA binding to TMD sites, which, due to their proximity, could directly influence the pore gate. Overall, the totality of benzo GABA effects at the canonical ECD site appears to be more intricate than the simplified approximation of solely modulating channel pre-activation.
9. Structural Insights into Benzo GABA Modulation in the ECD
Advances in cryo-EM have provided structural snapshots of heteromeric GABAA(α1)2(βX)2(γ2)1 receptors in complex with both agonists and benzo GABA [42,43,44,45,46,47,48]. These studies validate the canonical high-affinity benzo GABA site in the ECD, as identified by functional studies, and reveal details, along with subunit specificity, of lower-affinity benzo GABA sites in the TMD (Figure 1A–C). Combined with structures of homologous pentameric ligand-gated ion channels (pLGICs) like nAChRs, GlyRs, 5-HT3 receptors, and prokaryotic homologues [102], these structural snapshots generally align with the overall subunit motions during agonist gating. Comparison of agonist-bound structures with unliganded or antagonist-bound structures indicates that agonist binding is associated with a more compact ECD conformation, increased buried surface area at intersubunit interfaces, and inward movement of loop C over the agonist. Furthermore, each subunit’s ECD undergoes rotation and tilting, leading to expansion at the ECD/TMD interface, radial outward movement of M2-M3 linkers, and splaying apart of pore-lining M2 helices at their top. This splaying opens the hydrophobic 9′ leucine ring near their center, which forms the activation gate.
Comparisons of GABAA(α1)2(β2–3)2(γ2)1 receptor structures with and without classical benzo GABA like DZ reveal only subtle conformational differences in the ECD [44,45,46,103]. Based on this, Masiulis et al. proposed that benzo GABA PAMs, at their high-affinity site, stabilize the α+/γ− subunit interface. They suggested this stabilization aids in global ECD compaction during gating, thereby promoting channel opening [45]. In contrast to PAMs, occupation of the α+/γ− ECD site by the benzo GABA antagonist flumazenil induces a slight ECD expansion [103].
Supporting the idea that α+/γ− interface stability is crucial for benzo GABA modulation, cysteine cross-linking between α and γ subunits, in proximity to the critical histidine residue at the back of the canonical benzo GABA site in the α subunit β4–β5 linker, mimics benzo GABA action and is reversible upon disulfide bond reduction [104] (Figure 2ii). Conversely, glycine residue insertions to increase β4–β5 linker flexibility in α, β, or γ subunits all impair benzo GABA potentiation of GABA-evoked currents [105] (Figure 2iii). The importance of linkers in all subunits suggests that benzo GABA effects involve global conformational changes coupling benzo GABA and agonist binding sites via interactions between the β4–β5 linker at the back of each binding site and the neighboring subunit beta barrel. Indeed, mutations affecting benzo GABA modulation are not confined to a specific domain, supporting the notion of a more global effect on channel conformation.
Given the rigid body movement of subunit ECD beta barrels, dynamics are expected to occur primarily at intersubunit interfaces, including GABA and benzo GABA binding sites [106]. Only one GABA site shares the α subunit with the benzo GABA site, raising the possibility that benzo GABA may induce local changes preferentially modulating activation via a single GABA site. However, serial disruption of each GABA binding site in concatenated receptors revealed that benzo GABA can modulate currents in response to GABA binding at either site [107]. Beyond benzo GABA- and agonist-binding α+/γ− and β+/α− interfaces, epilepsy-related mutations disrupting electrostatic interactions at the nonbinding γ+/β− interface reduce benzo GABA-receptor complex stability [108]. Thus, destabilization (or stabilization) of one interface can lead to more global destabilization (or stabilization) at other interfaces, consistent with benzo GABA influencing the stability of intersubunit contacts throughout the ECD.
While benzo GABA likely stabilizes the α+/γ− interface, it’s important to note that all structures of benzo GABA-bound GABAA(α1)2(β2–3)2(γ2)1 receptors to date were obtained with both agonist sites occupied by GABA. The inability of benzo GABA to potentiate currents evoked by saturating GABA is hypothesized to reflect a near-complete biasing of the receptor to a pre-active conformation by the larger energetic contribution from both bound agonists, effectively overshadowing the smaller contribution from ECD benzo GABA binding. Thus, the lack of significant ECD motions associated with benzo GABA in these conditions is perhaps not surprising. Structures of benzo GABA complexes with zero or one bound agonist, or with partial agonists, which might better reveal motions associated with benzo GABA binding, are currently lacking.
Structural models of homologous GlyR and ACh binding protein in complex with partial agonists suggest that the initial pre-activation step involves loop C swinging towards the bound agonist to adopt a more compact conformation [109,110]. While the styrene maleic acid (SMA) copolymer used to solubilize GlyR in this study may have introduced conformational bias, this bias may have fortuitously aided the observation of a normally transient pre-active state. Considering these GlyR/ACh binding protein models alongside GABAA(α1)2(βX)2(γ2)1 receptor structures suggests a hypothetical mechanism for benzo GABA modulation via the canonical high-affinity site: benzo GABA PAMs promote a pre-activated, more compact ECD conformation with loop C closed more tightly around the binding pocket by stabilizing or enhancing global intersubunit interactions. It’s reasonable to assume that NAMs might exert the opposite effect by destabilizing intersubunit interfaces. Consistent with this, GABAA(α1)2(β2)2(γ2)1 receptor structures complexed with a benzo GABA antagonist or NAM exhibit a less compact ECD compared to complexes with PAMs [46,48].
10. Structural Mechanisms of Benzo GABA Modulation in the TMD
To ensure binding site saturation, GABAA(α1)2(βX)2(γ2)1 structures were solved in high micromolar DZ concentrations, sufficient to populate lower-affinity TMD sites. The observed TMD binding sites validate binding residues identified through mutagenesis and highlight asymmetric binding to specific subunit interfaces [46,63]. DZ binds within an intersubunit pocket between TM helices of β and α subunits, directly below GABA binding sites, and in a homologous pocket at the γ+/β− interface. Pore-lining M2 helices contribute to these binding pockets, through which DZ may directly influence pore opening. Interestingly, these TM sites are identical to those binding anesthetics like propofol at the β+/α− interface and barbiturates like phenobarbital at the γ+/β− interface [46,62,111]. Therefore, lower-affinity binding to these sites may contribute to the anesthetic effects observed at high benzo GABA concentrations, such as DZ. At nanomolar to low micromolar DZ concentrations, commonly used in functional studies, the observed effects are likely primarily due to binding at the canonical high-affinity ECD site. Nevertheless, in the absence of definitive evidence to the contrary, binding at TM sites in a subset of receptors could complicate data interpretation.
In contrast to the subtle benzo GABA-associated motions in the ECD, benzo GABA exerts more pronounced effects on TMD structure. Diazepam binding in the TMD results in global TMD stabilization, with tighter packing of TM helices from neighboring subunits. Strikingly, while the benzo GABA antagonist flumazenil doesn’t bind in the TMD, its binding to the canonical ECD site destabilizes the TMD, increasing the gap between neighboring subunit TM helices. This suggests that benzo GABA binding to the canonical ECD site can induce structural changes within the TMD. Consistent with this, the accessibility of TM domains to MTS reagents is altered by benzo GABA, an effect that is asymmetric across subunits [112,113].
While TMD stability modulation by benzo GABA has been observed by one research group [46,103], another group reported minimal TMD changes [44,45]. One explanation for this discrepancy could be that tighter wrapping of the nanodisc scaffold in the latter group’s preparation constrained TMD motions and prevented benzo GABA-induced changes. Alternatively, truncation of the intracellular M3-M4 loop by the former group may have resulted in an abnormally destabilized TMD. Both groups utilized nanobodies bound to the ECD to aid molecule orientation determination, which could also influence ECD conformations. The extent to which these experimental preparations influenced the resulting structures is a crucial question for interpreting structural mechanisms that remain to be fully understood.
11. Elucidating Benzo GABA-to-Pore Coupling Mechanisms
While the general concept of benzo GABA, like DZ, stabilizing a more compact pre-active receptor conformation with enhanced intersubunit interface interactions is supported by diverse experimental evidence, the specific interactions and microdomain motions critical for benzo GABA modulation remain to be fully elucidated. For example, cysteine cross-linking between the γ subunit β8–β9 loop (beneath the benzo GABA binding site) and either the neighboring β9 strand in the same subunit or the β1–β2 loop in the adjacent α subunit impairs modulation by the benzo GABA flurazepam. This suggests that a more flexible β8–β9 loop is important for benzo GABA’s modulatory effects [114] (Figure 2iv–vi). Conversely, cross-linking the β9 loop to the neighboring pre-M1 region, connecting the ECD to the M1 helix, enhances flurazepam modulation (Figure 2vii). The γ subunit β8-β9 loop extends along the bottom outer edge of the benzo GABA binding pocket towards the intersubunit region near the ECD/TMD interface. Here, it can interact with the M2-M3 linker, Cys-loop, and β1–β2 loop in the neighboring α subunit – a region known to be crucial for channel activation [115,116] (Figure 2). This loop has been implicated in both transducing modulatory effects and benzo GABA binding [57,60,117,118,119]. The homologous loop in the α subunit has also been implicated in GABA-mediated gating [120] and was observed to undergo significant motions upon activation of the prokaryotic homologue GLIC [121].
A challenge in interpreting studies examining benzo GABA modulation of GABA-evoked currents lies in distinguishing between direct pore gating changes and indirect alterations in the pore closed–open equilibrium resulting from GABA affinity changes. A method to differentiate these effects is to measure the energetic perturbation benzo GABA binding exerts on the pore gate in the absence of GABA. Spontaneously active mutants, whose closed–open equilibrium is sensitive to benzo GABA alone, enable this approach. Using this strategy, a single valine residue at the center of the α1 subunit M2-M3 linker was identified. Mutation of this valine to alanine increases the efficiency of chemical energy transduction from DZ binding to the pore gate approximately threefold [88] (Figure 2viii). The reduced sidechain volume in the alanine substitution may contribute to this effect. Consistent with this, a mutation to a bulkier tryptophan did not enhance DZ efficiency, suggesting that a more flexible M2-M3 linker may also promote benzo GABA modulation, possibly through interactions with the neighboring γ subunit pre-M1 region or β8-β9 loop. How these observations relate to a more globally compact receptor conformation remains to be fully understood, as does the symmetry of mechanisms for PAMs and NAMs, where perturbations don’t always exhibit mirrored effects [114].
12. Conclusions and Future Directions in Benzo GABA Research
Despite substantial research efforts to unravel the basis of benzo GABA modulation, a complete molecular-level understanding of their mechanism of action remains elusive. While numerous studies have provided valuable insights and highlighted a role in facilitating receptor pre-activation, significant questions persist. From a structural perspective, three-dimensional information on benzo GABA-bound receptor conformations under conditions where benzo GABA exerts functional effects (e.g., with partial agonists or a single bound agonist) is lacking. For benzo GABA drugs like DZ, which bind in both ECD and TMD, structures with only one domain occupied would aid in dissecting the distinct contributions of binding in each domain. More broadly, a structure of a heteromeric GABAAR with a conducting pore is still needed, as all agonist-bound structures to date are in a putative desensitized state with pore constriction at the bottom of M2 helices [122,123]. Obtaining these structures likely presents a significant challenge due to the transient nature of pre-active and open conformations, causing agonist-bound receptors to rapidly accumulate in desensitized states. Furthermore, a clearer understanding of how solubilization scaffolds or nanobodies influence channel conformation is crucial for reliable interpretation of structural observations. Functionally, further research is needed to dissect the detailed molecular interactions involved in benzo GABA modulation. Utilizing gain-of-function mutants that enable unambiguous measurement of benzo GABA-to-pore coupling energetics, without the complexities of agonist binding, may prove particularly fruitful in this regard. Thus, while a general understanding of benzo GABA modulation mechanisms is emerging, much remains to be discovered to fully elucidate the physical basis of this important class of psychotropic modulators, specifically how benzo GABA interacts with the GABAA receptor to produce its therapeutic effects.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Funding Statement
Department of Neuroscience Startup, University of Texas at Austin.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.